摘要:日本名古屋工业大学(Nagoya Institute of Technology)Norio Shibata课题组发展了首例硅烷基硼酸酯介导的有机氟化物与二级胺的脱氟偶联反应,在室温下即可实现C-F键与N-H键的偶联
近日,日本名古屋工业大学(Nagoya Institute of Technology)Norio Shibata课题组发展了首例硅烷基硼酸酯介导的有机氟化物与二级胺的脱氟偶联反应,在室温下即可实现C-F键与N-H键的偶联。此反应条件温和,有效避免了热力学诱导的SN2或SN1胺化。此转化的优势在于在硅烷基硼酸酯介导下可以选择性的实现有机氟化物的C-F键活化,而分子内的C-O、C-Cl、杂芳基C-H、C-N以及CF3均不受影响。此转化具有良好的底物适用性和官能团兼容性,一步高效实现了一系列(杂)芳基三级胺、脂肪三级胺的高效合成,该方法还可用于候选药物包括其氘标记类似物的后期合成。相关成果发表在Nat. Commun.上,文章链接DOI:10.1038/s41467-023-37466-0。
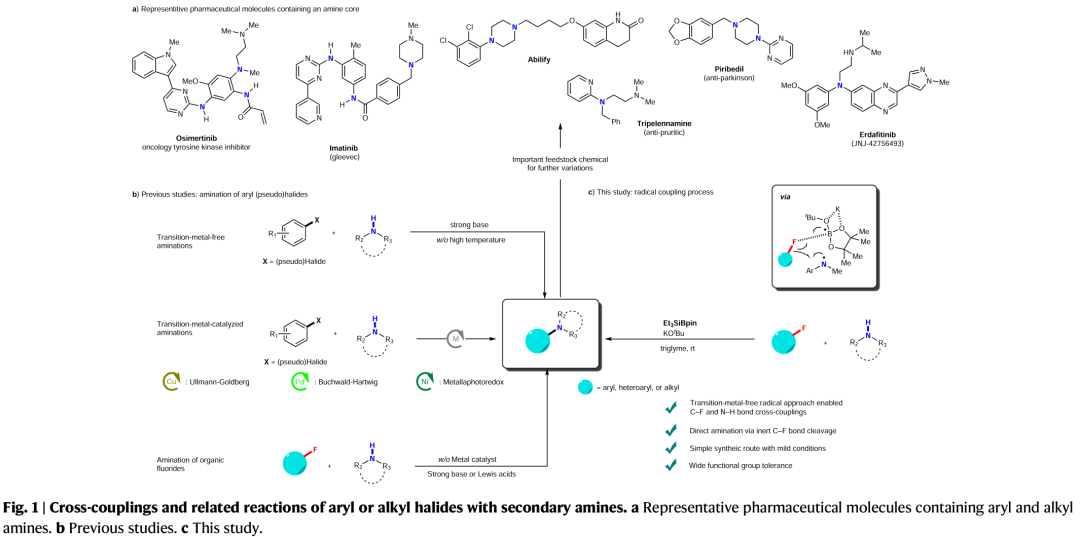
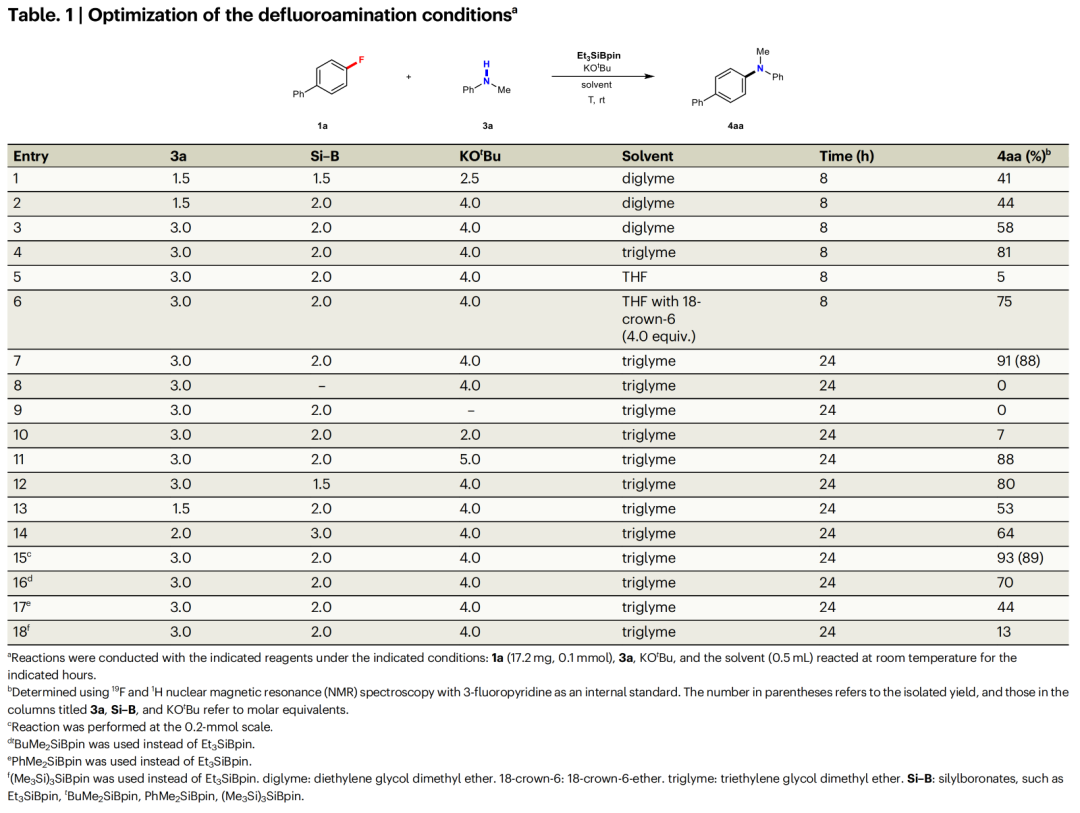
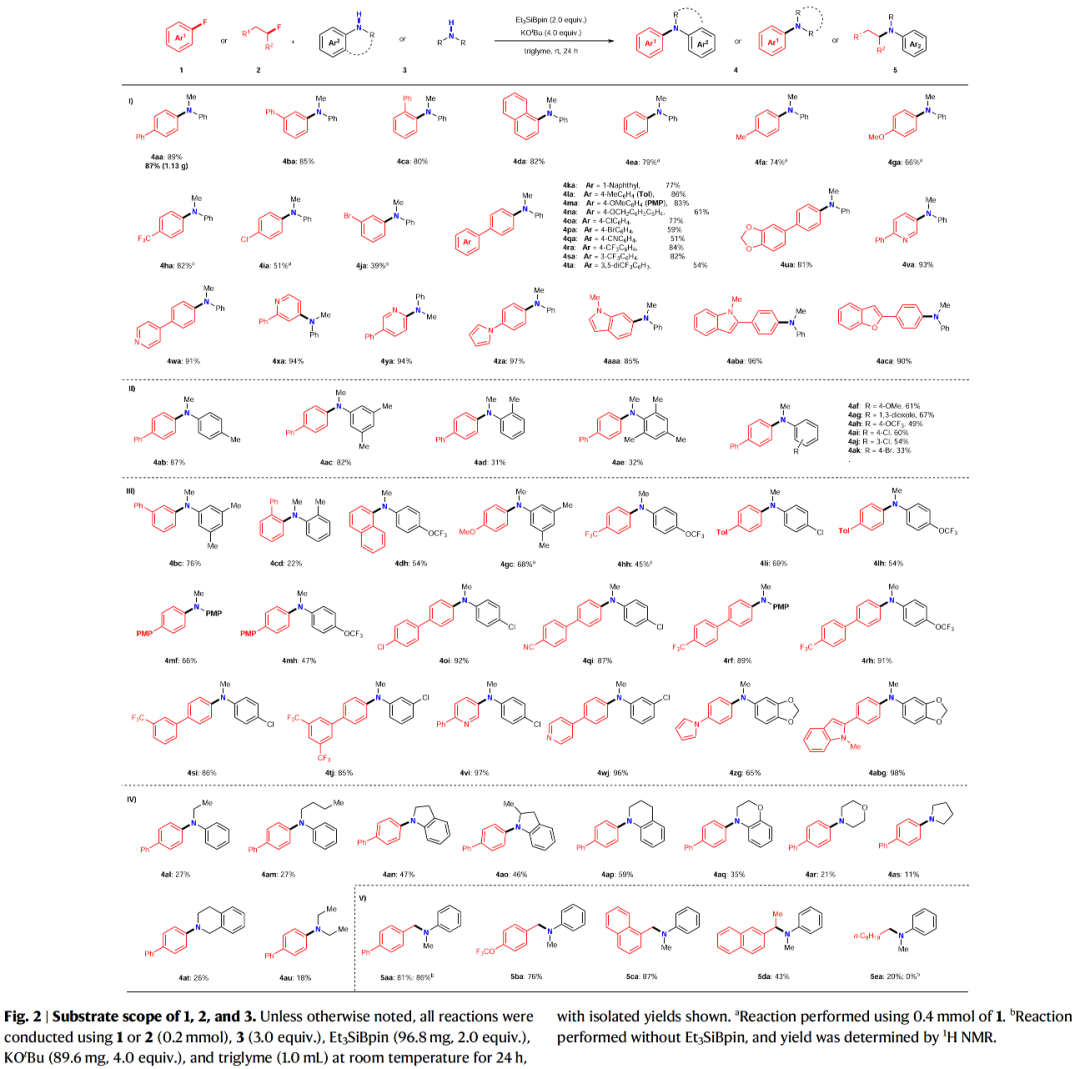
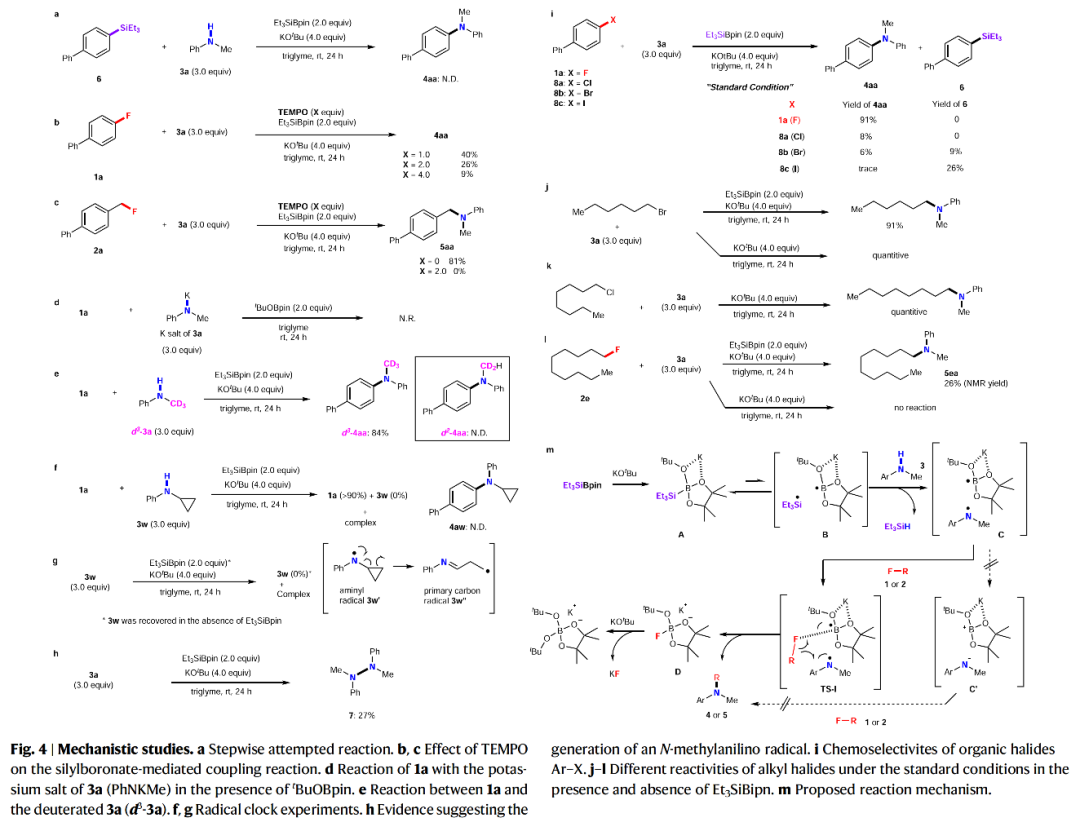
含氟有机化合物广泛存在于药物和农用化学品中,因为氟原子的引入可以很好的调节此类化合物的化学稳定性和代谢稳定性、亲脂性以及酸/碱度。然而,实现含氟有机化合物的C-F键的合成转化则具有一定的挑战性。与C-I/Br/Cl键相比,C-F键更加惰性且具有最高的键解离能。因此实现C-F键的断裂通常需要强碱来促进反应,并且需要较高的反应温度。芳香叔胺广泛存在于制药、农业科学、生物活性天然产物和材料科学的相关分子中(Fig. 1a)。迄今为止,芳香叔胺最可靠的制备方法是过渡金属催化芳基卤化物与胺亲核试剂的C(sp2)-N偶联,如Ullmann偶联、Buchwald–Hartwig反应、金属光氧化还原胺化等(Fig. 1b)。尽管过渡金属催化的胺化反应已经取得了一定的进展,但是其通常具有区域选择性差、需要使用强碱、反应温度高等不足。此外,过渡金属催化剂的使用不符合当今绿色化学的理念。最近,日本名古屋工业大学Norio Shibata课题组发展了首例硅烷基硼酸酯介导的有机氟化物与二级胺的脱氟偶联反应,在室温下即可实现C-F键与N-H键的偶联。此反应条件温和,有效避免了热力学诱导的SN2或SN1胺化。(Fig. 1c)。下载化学加APP到你手机,更加方便,更多收获。首先,作者以4-氟联苯1a和N-甲基苯胺3a作为模板底物进行反应探索(Table 1),经过一系列条件筛选,作者发现当使用1a (0.1 mmol), 3a (3.0 equiv.), Et3SiBpin (2.0 equiv.), KOtBu (4.0 equiv.),在三甘醇二甲醚(triglyme)中室温反应24 h,可以以89%的分离产率的到产物4aa(Table 1, entry 15)。在得到了最优条件后,作者对此转化的底物范围进行了探索(Fig. 2)。实验结果表明,此转化对一系列不同取代的芳基氟化物和二级胺均具有良好的兼容性,以中等至良好的产率实现了产物4aa-4aca, 4ab-4ak, 4bc-4abg, 4al-4au的合成。此外,此转化对一系列烷基氟化物也具有良好的适用性,以20-87%的产率得到产物5aa-5ea。值得注意的是此体系对卤素、烷基、三氟甲氧基、三氟甲基、甲氧基、氰基、杂芳基、芳基等一系列官能团均可兼容,展现了此体系的良好应用性。接下来,为了证明此转化的应用性,作者应用此体系进行了一系列合成转化(Fig. 3)。对于(±)-α-tocopherol衍生的芳基氟化物1ad、(-)-menthol衍生的N-甲基苯胺3v、estrone衍生物1ae均可顺利参与反应,分别得到相应的产物4ada(83%)、4av(75%)和4aev(62%)。此外,鉴于N-烷基骨架和C-D键在药物化学研究中的重要性,作者利用d3-3a与生物活性分子衍生物1ad-1ag反应,可以以70-88%的产率得到相应的含N-CD3产物d3-4ada-d3-4aga。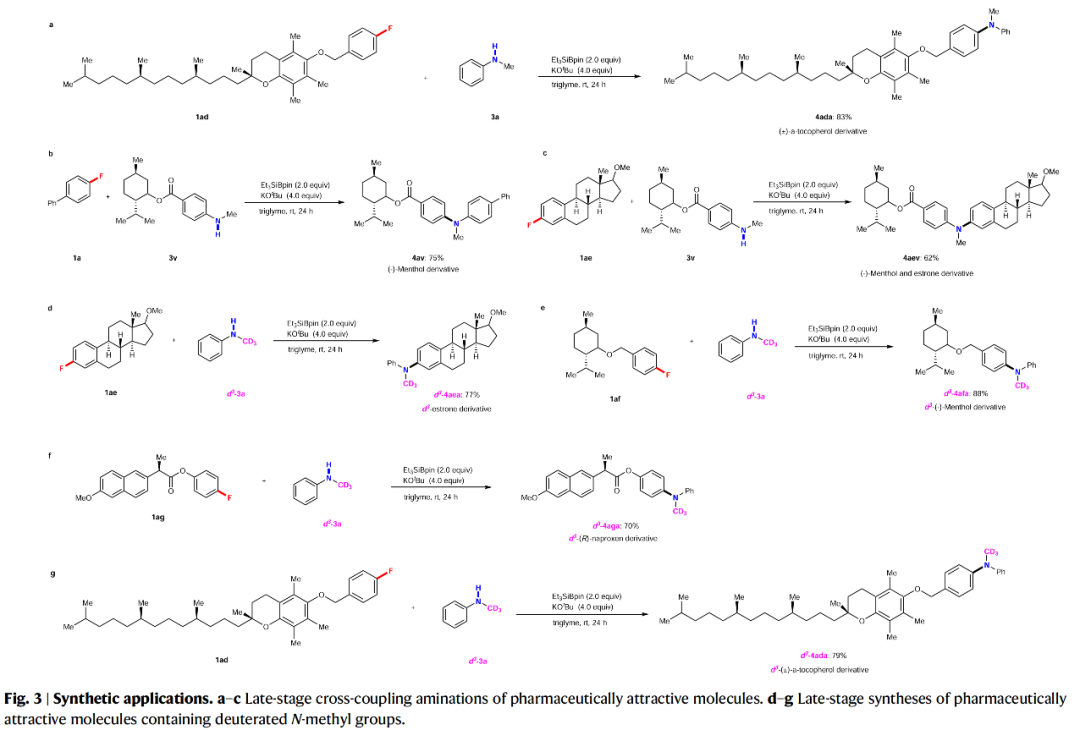 为了深入理解反应机理,作者进行了一系列控制实验(Fig. 4)。当使用硅基化合物6与3a在最优条件下反应时,并没有检测到产物4aa,由此表明6不能够参与反应(Fig. 4a);当在1a的反应体系中加入4.0 equiv TEMPO时,仅以9%的产率得到产物4aa(Fig. 4b);当在2a的反应体系中加入2.0 equiv TEMPO时,并未观察到产物5aa(Fig. 4c);当1a与新制备的3a钾盐在tBuOBpin存在下并没有反应发生,由此表明反应中并不包含苯胺钾盐的形成(Fig. 4d);当使用d3-3a在最优条件下反应时,可以以84%的产率分离到产物d3-4aa,而并未得到d2-4aa,由此表明反应中并不涉及底物的N-甲基部分的转化(Fig. 4e);利用1a与3w的自由基钟实验得出1a几乎完全回收,而3w则转化成了复杂的混合物(Fig. 4f);此外,仅使用3w在标准条件下反应也得到相同的混合物。而在体系中不存在Et3SiBPin时,3w可以回收(Fig. 4g);当利用3a在不存在1a的条件下反应时,可以以27%的产率观察到肼7的形成,可能是通过N-甲基苯胺自由基二聚所形成的(Fig. 4h)。上述控制实验表明此脱氟C-N偶联过程是经历自由基路径进行的。接下来,作者通过对传统的芳基卤化物 Ar-X 8a-c (X = Cl, Br, or I)与芳基氟化物1的反应进行比对,得出此体系对C-F键的转化具有特定的化学选择性(Fig. 4i)。随后,作者对不同烷基卤化物的活性进行比对,得出烷基溴化物(Fig. 4j)和氯化物(Fig. 4k)无论在Et3SiBpin存在与否均可以较高的产率得到胺化产物。而烷基氟化物在Et3SiBpin存在下仅以26%的产率得到产物5ea,并且在不存在Et3SiBpin时候反应是不发生的(Fig. 4l)。上述控制实验表明C-F键与C-X(Cl, Br)键的断裂相比更具有挑战性。基于上述实验结果,作者提出了此转化可能的反应机理(Fig. 4m): 首先,Et3SiBpin与KOtBu反应形成中间体A;随后,由于-OtBu的单电子还原性质,中间产物A分裂成由三乙基硅基自由基(•SiEt3)和硼自由基(B•)组成的自由基对B;接着•SiEt3从3中攫氢得到自由基对C并形成HSiEt3;接下来,自由基对C吸引有机氟化物1或2得到TS-I;最后,TS-I通过C-N键的形成得到目标产物4或5,并释放D ([Bpin(OtBu)F]K,而D 可以与KOtBu反应生成稳定的[Bpin(OtBu)2]物种和KF。
为了深入理解反应机理,作者进行了一系列控制实验(Fig. 4)。当使用硅基化合物6与3a在最优条件下反应时,并没有检测到产物4aa,由此表明6不能够参与反应(Fig. 4a);当在1a的反应体系中加入4.0 equiv TEMPO时,仅以9%的产率得到产物4aa(Fig. 4b);当在2a的反应体系中加入2.0 equiv TEMPO时,并未观察到产物5aa(Fig. 4c);当1a与新制备的3a钾盐在tBuOBpin存在下并没有反应发生,由此表明反应中并不包含苯胺钾盐的形成(Fig. 4d);当使用d3-3a在最优条件下反应时,可以以84%的产率分离到产物d3-4aa,而并未得到d2-4aa,由此表明反应中并不涉及底物的N-甲基部分的转化(Fig. 4e);利用1a与3w的自由基钟实验得出1a几乎完全回收,而3w则转化成了复杂的混合物(Fig. 4f);此外,仅使用3w在标准条件下反应也得到相同的混合物。而在体系中不存在Et3SiBPin时,3w可以回收(Fig. 4g);当利用3a在不存在1a的条件下反应时,可以以27%的产率观察到肼7的形成,可能是通过N-甲基苯胺自由基二聚所形成的(Fig. 4h)。上述控制实验表明此脱氟C-N偶联过程是经历自由基路径进行的。接下来,作者通过对传统的芳基卤化物 Ar-X 8a-c (X = Cl, Br, or I)与芳基氟化物1的反应进行比对,得出此体系对C-F键的转化具有特定的化学选择性(Fig. 4i)。随后,作者对不同烷基卤化物的活性进行比对,得出烷基溴化物(Fig. 4j)和氯化物(Fig. 4k)无论在Et3SiBpin存在与否均可以较高的产率得到胺化产物。而烷基氟化物在Et3SiBpin存在下仅以26%的产率得到产物5ea,并且在不存在Et3SiBpin时候反应是不发生的(Fig. 4l)。上述控制实验表明C-F键与C-X(Cl, Br)键的断裂相比更具有挑战性。基于上述实验结果,作者提出了此转化可能的反应机理(Fig. 4m): 首先,Et3SiBpin与KOtBu反应形成中间体A;随后,由于-OtBu的单电子还原性质,中间产物A分裂成由三乙基硅基自由基(•SiEt3)和硼自由基(B•)组成的自由基对B;接着•SiEt3从3中攫氢得到自由基对C并形成HSiEt3;接下来,自由基对C吸引有机氟化物1或2得到TS-I;最后,TS-I通过C-N键的形成得到目标产物4或5,并释放D ([Bpin(OtBu)F]K,而D 可以与KOtBu反应生成稳定的[Bpin(OtBu)2]物种和KF。(图片来源:Nat. Commun.)
总结
Norio Shibata课题组在室温下通过惰性C-F键活化开发了首例硅烷基硼酸酯介导的有机氟化物与二级胺的自由基偶联。多种二级无环和环状N-烷基苯胺和二烷基胺都可与不同的有机氟化物反应,在非常温和的条件下以中等至优异的收率生成结构多样的芳香族叔胺,该方案可兼容含有OR-、Cl-、CN-或CF3官能团的反应物。此外,该方法避免使用过渡金属和专用配体。该方法还可以兼容一系列生物活性分子衍生物,具有良好的应用性。