JACS:水作氧化剂,常温下甲烷和CO直接光催化合成乙酸
2023-02-15
摘要:法国里尔大学安德烈· Y· Khodakov和维塔利 V.奥多姆斯基小组报告了一条可以在常温下驱动CH4, CO,水合成乙酸的新型光催化方法。在光催化作用下,能在60小时内稳定地制备出5.7 mmol·L–1的醋酸溶液
法国里尔大学安德烈· Y· Khodakov和维塔利 V.奥多姆斯基小组报告了一条可以在常温下驱动CH4, CO,水合成乙酸的新型光催化方法。在光催化作用下,在 Pt/W/Pt/PW/TiO2 (Pt/W-NPW)/TiO2的作用下,能在60小时内稳定地制备出5.7 mmol·L–1的醋酸溶液,
在液相和碳基础上对乙酸分别具有90%和66%以上的选择性,每摩尔Pt产生99 mol乙酸。同位素和原位光谱表明,乙酸合成是通过Pt1位点上CH4的光催化氧化羰基化进行的,CH4的活化由水衍生的羟基自由基促进
甲烷作为温室气体和最丰富的碳质储备原料之一,将其催化转化为高附加值的液态含氧化合物引起研究者极大的兴趣。目前,CH4转化为乙酸(AcOH)的工业路线是一条能源密集型间接多步骤路线,首先CH4转化为合成气、甲醇合成,随后甲醇羰基化为AcOH。虽然已有研究表明在温和条件下可以将甲烷直接转化为AcOH,但反应中加入的强氧化剂(如腐蚀性酸或分子氧)会导致环境问题、安全风险和低选择性。因此,在避免使用腐蚀性化学品的同时,实现CH4的温和选择性氧化为高浓度AcOH将是最佳方案。
Sushkevich等人表明在CH4无氧氧化中水作为软氧化剂和中间稳定剂有助于甲烷的高选择性,Liu等人进一步阐述水作为O提供者和位点阻断剂促进了CH4氧化为甲醇(Science, 2020, 368, 513-517)。但将CH4、CO和水直接合成AcOH的挑战主要是其不利的热力学(Equations 1,2)。
CH4 + CO + H2O = CH3COOH + H2 (1)
2CH4 + 2H2O = CH3COOH + 4H2 (2)
光催化技术可以将光激发的电荷载体作为化学键活化的驱动力,完成室温下水的活化,因此上述H2O诱导CH4氧化转化为AcOH是可行和可持续的。
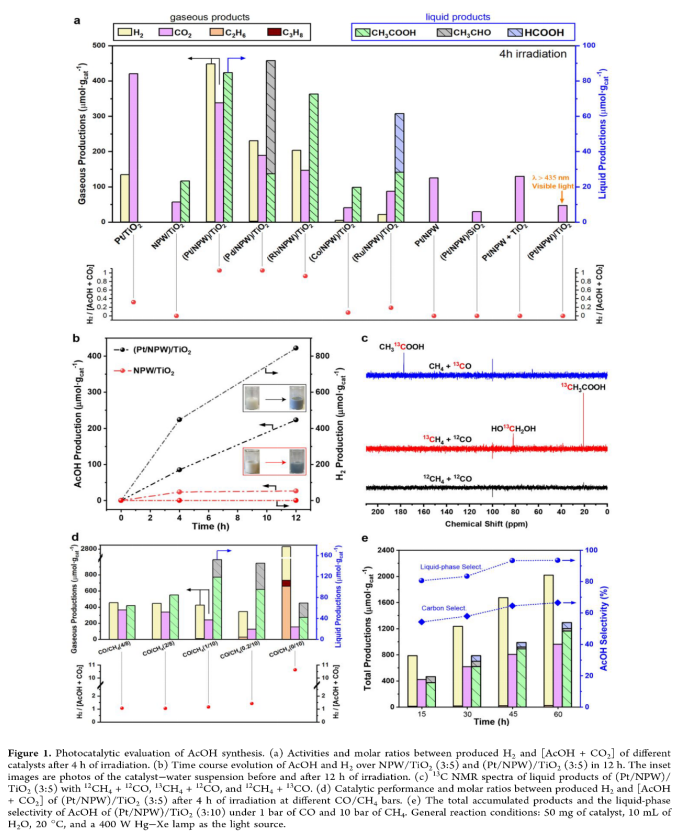
Figure 1a所示为不同光催化剂在2 bar CO和8 bar CH4下光照反应4 h的不同产物产率。Pt/TiO2只能将CO和/或CH4转化为CO2和H2,而不产生有价值的液态含氧化合物。仅在经过研磨和热处理的(M/NPW)/TiO2(M=Pt、Pd、Rh、Co和Ru)和NPW/TiO2中观察到AcOH的生成,且(Pt/NPW)/TiO2显示出最高的活性,AcOH产率为84.7 μmol·gcat-1。当可见光(λ>435 nm)照射(Pt/NPW)/TiO2时,仅产生非常少量的CO2,Pt/NPW和(Pt/NPW)/SiO2也表现出类似光催化行为。这些结果表明(Pt/NFW)/TiO2上的AcOH合成基本上由TiO2的光诱导电荷载流子驱动。Figure 1b表明(Pt/NPW)/TiO2可稳定生成AcOH,NPW/TiO2仅在初始阶段观察到AcOH的生成。由于部分还原NPW(即杂多蓝)的形成,两种催化剂在光照12 h后颜色都变为深蓝色(inset images)。这表明甲烷在NPW/TiO2和(Pt/NPW)/TiO2上的氧化可能涉及NPW晶格氧:
CH4 + CO + NPW = CH3COOH + NPW[Ov] (3)
这些氧物种的耗尽可能导致NPW/TiO2的失活。但将失活的NPW/TiO2暴露在空气中搅拌2 h,可以在下一个循环中恢复其颜色和反应性。
(图片来源:J. Am. Chem. Soc.)
作者研究了CO/CH4比率对(Pt/NPW)/TiO2催化性能的影响。如Figure 1d所示,1bar CO和10 bar CH4下,光照4 h后,AcOH产率达到118.5 μmol·gcat-1。高生产率伴随着额外的乙醛生产,对AcOH的液相选择性为78%。CO分压从4 bar降低到0.2 bar后CO2产量显著降低,CO2和AcOH的总量约等于混合CO/CH4气氛下H2的产量(图1d)。在10 bar CH4下,产生大量C2H6、C3H8和H2,表明非氧化甲烷与乙烷的偶联成为主要反应路径;同时AcOH产量减少了3倍(Figure 1d)。在没有添加CO的情况下,甲烷产生少量AcOH可能是由于甲烷部分氧化过程中产生的甲烷和CO的耦合。
作者进一步优化催化剂结构,当Pt负载量为0.2 wt%,Pt/NPW与TiO2的重量比为3:10时,催化剂表现出最高的AcOH产率(374.4 μmol·gcat-1)和2.55%的表观量子产率。优化的(Pt/NPW)/TiO2(3:10)催化剂表现出优异的光化学稳定性和选择性(Figure 1e)。在1 bar CO和10 bar CH4下,反应60 h、四个循环反应后,产生1166 μmol·gcat-1 AcOH(无再生处理)。四个循环反应的AcOH产率依次为24.9、16.5、17.9和18.4 μmol·h-1gcat-1。假设活性位点为Pt位点,反应TON为99,AcOH水溶液浓度为5.7 mmol·L-1。
(图片来源:J. Am. Chem. Soc.)
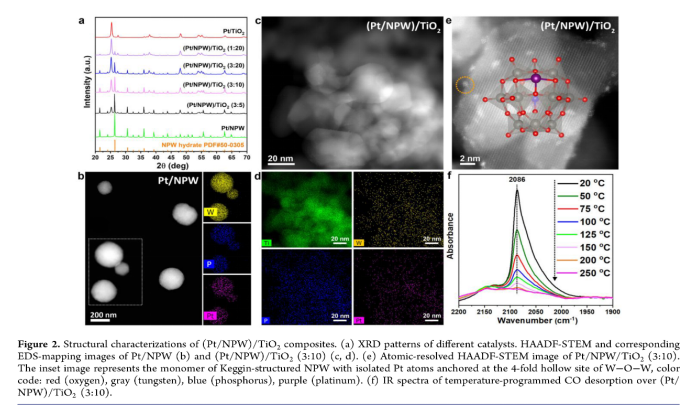
为了进一步了解产AcOH的催化活性位点,作者对(Pt/NPW)/TiO2结构进行研究。纯Pt/NPW XRD中Keggin型NPW晶相的窄衍射峰表明Pt/NPW的晶粒尺寸较大。(Pt/NPW)/TiO2由TiO2和Keggin型NPW晶相构成,随着(Pt/NPW)含量的降低,NPW信号的强度相应降低,直到(Pt/NFW)/TiO2比例为1:20时NPW XRD峰几乎完全消失(Figure 2a)。如Figure 2b所示,STEM图显示Pt/NPW是直径为几百纳米的大型多面体颗粒,高角度环形暗场(HAADF)成像无法区分Pt和W原子量。EDS图揭示样品中Pt物种均匀分散在单个NPW多面体上。(Pt/NPW)/TiO2(复合物比例3:10) STEM和EDS图显示W、P和Pt物种高度分散在TiO2上(Figure 2c,d),HAADF成像显示这些高度分散的物种呈孤立的Keggin型NPW簇(Figure 2e)。利用CO的程序升温脱附(TPD)研究(Pt/NPW)/TiO2(3:10)中Pt的分散情况,红外光谱(IR)表征表面物种。在0.1 bar CO和0.9 bar CH4,紫外光照射下原位预处理催化剂。照射60 min观察到对应Pt物种上CO吸附的2086 cm-1中心强吸收带(Figure 2f)。在黑暗、真空下,温度从20 °C升到250 °C, CO从催化剂中逐渐脱附。随着CO覆盖率的降低,CO的峰值始终保持在2086 cm-1,表明吸附的CO分子之间没有偶极-偶极相互作用,并证实Pt物种在催化剂上的原子分散。
(图片来源:J. Am. Chem. Soc.)
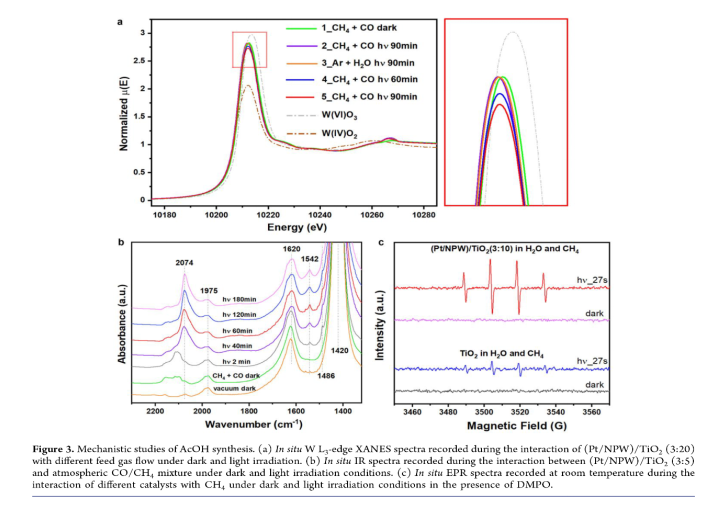
作者结合原位光谱和同位素方法,研究了CH4、CO和H2O光催化合成AcOH的机理。在CH4/CO和Ar/H2O中对(Pt/NPW)/TiO2(3:20)进行光照射时,X-射线吸收近边缘结构(XANES)光谱显示白线边缘发生变化(光谱2,3),在催化剂长时间暴露于CH4/CO(光谱4,5)后白线强度略有降低,表明W物种逐渐减少(Figure 3a)。这归因于CH4/CO中W物种的部分还原。同位素标记试验表明AcOH的甲基来源于CH4,酰基来源于CO。作者通过原位FTIR对光照下(Pt/NPW)/TiO2(3:5)表面中间体的转化进行监测(Figure 3b)。在真空、黑暗状态下,1420和1620 cm-1处峰值分别对应于NPW中NH4+和吸附H2O的振动。在0.1 bar CO和0.9 bar CH4下,2150-2100 cm-1的峰值对应于阳离子Pt物种上吸附CO的峰。光照后,2074 cm-1处Pt1位点上CO吸附峰发生变化,并在1486和1542 cm-1处形成新峰,这归因于*CH3COO的对称(νs)和不对称(νas)振动。随着光照时间延长,νas(*CH3COO)峰变强,1620 cm-1处峰强降低(对应于光催化反应吸收的H2O的消耗)。原位EPR表明催化剂在光照下生成羟基自由基(•OH) (Figure 3c)。
(图片来源:J. Am. Chem. Soc.)
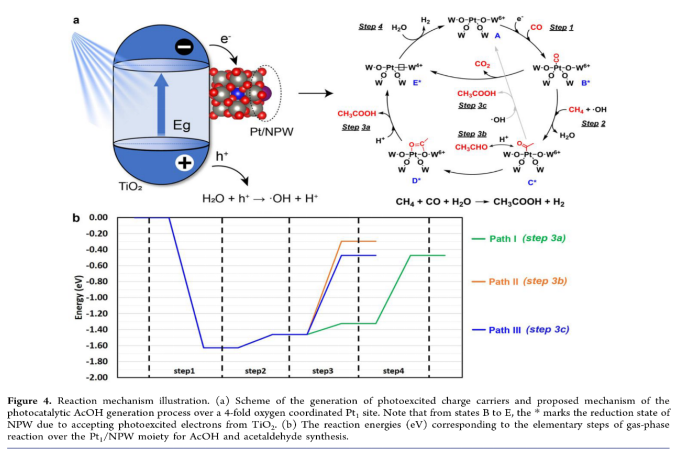
Figure 4a所示为CH4、CO和水催化产AcOH机制。UV照射后TiO2激发电子迁移到Pt/NPW簇表面,空穴保留在TiO2上与H2O反应生成H质子(H+)和•OH。FTIR数据表明CO吸附优先发生在Pt1位点(步骤1)。甲烷在Pt1位点上被•OH活化,并通过FTIR检测形成乙酰基,该乙酰基由CH4与吸附的CO的C−C偶联形成(步骤2)。乙酰基氧化成乙酸盐并作为AcOH脱附(步骤3a)。AcOH脱附在Pt1位点附近产生氧空位,并导致W6+还原为W4+。这些氧空位由H2O分子中的氧填充,产生H2并将W4+再氧化回W6+(步骤4)。催化反应涉及分散在TiO2上的Pt/NPW晶格氧物种(路径I)。AcOH也可以通过乙醛中间体的氧化(路径II中的步骤3b)或乙酰基被•OH直接氧化(路径III中的步骤3c)产生。Figure 4b理论计算表明,由于较高的吸热性,与AcOH相比,路径II形成乙醛是不太有利的途径。路径I和III表现出类似的能量来完成反应循环。
Maxwell J. Robb团队报道了一种新的光催化途经,在环境温度下,通过(Pt/NPW)/TiO2将CH4、CO和H2O直接光催化合成AcOH。在优化的条件下,催化剂能够在60 h内稳定合成5.7 mmol·L-1 AcOH溶液,液相选择性超过90%,在光生载流子驱动下,CH4被•OH自由基激活并与吸附在Pt1表面位点上的CO分子反应,产生乙酰基,乙酰基进一步氧化生成乙酸
相关阅读




